
Translational Research in Hereditary Spastic Paraplegia
By Darius Ebrahimi-Fakhari, MD, PhD
Boston Children’s Hospital
The hereditary spastic paraplegias (HSPs) are a group of over 80 rare or ultra-rare genetic diseases that collectively represent the most common cause of inherited spasticity and associated disability worldwide, with an estimated prevalence of 2-5:100,000. All HSPs present with progressive degeneration of the long axonal tracts, leading to significant motor impairment. There are no therapies that halt disease progression, highlighting the urgency for research into the fundamental biology of HSP.
AP-4-associated forms of hereditary spastic paraplegia
In 2018, our team was introduced to two little girls with an ultra-rare form of HSP, hereditary spastic paraplegia type 47 (SPG47). These two families had met in our Neurology Clinic and connected with our Translational Neuroscience Center. At the time, there were less than 10 patients reported with this condition; none in North America.
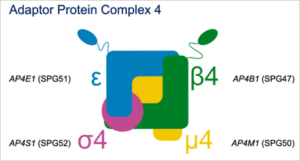
Figure !
SPG47 is one of four diseases causes by bi-allelic loss-of-function variants in the same protein complex, the adaptor protein complex 4 (or AP-4) (Figure 1). The AP-4 complex is an obligate heterotetramer and loss of any of the subunit proteins leads to the same molecular problem – loss of AP-4 function, and, as we know now, the same set of symptoms.
Research Goals
Our research is focused on two key areas:
- To create clinical trial readiness through a natural history study and biomarker discovery.
- To work towards molecular therapies including gene replacement strategies (with collaborators) and high-throughput small molecule screens for drug repurposing and drug discovery.

Figure 2
With the input of global collaborators, we created an international disease registry and natural history study for AP-4-associated hereditary spastic paraplegia (AP-4-HSP, ClinicalTrials.gov Identifier: NCT04712812). Many more children are affected by AP-4-HSP than we had initially realized. Over the years we have had the opportunity to meet with over 200 families (Figure 2).
Common Traits Discovered
Presenting Symptoms
A first cross-sectional analysis of over 150 children and young adults established that all four forms of AP-4-HSP share a common phenotype consisting of a core set of symptoms: Global developmental delay with prominent motor- and speech delays are usually the first symptoms, followed by onset of progressive spasticity of the legs, epilepsy, movement disorders and motor decline.
All children with AP-4-HSP present with global developmental delay and later intellectual disability that ranges from moderate to severe.
Additionally, we learned that many patients received a diagnosis of cerebral palsy before progressive clinical features were recognized and/or genetic testing was pursued.
Common Brain Features
A systematic assessment of brain MR imaging studies, led us to establish a set of core imaging features that now serve as a diagnostic imaging signature. These studies also showed that developmental brain malformations were common, including the underdevelopment of the corpus callosum and anterior commissure, as well as neuronal migration deficits leading to polymicrogyria.
Neurodevelopmental and a Neurodegenerative Disease
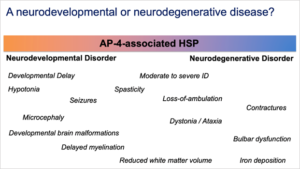
Figure 3
Our discoveries on the clinical and neuroimaging spectrum of AP-4-HSP has not only allowed us to counsel families more accurately, but has also enabled us to:
- gain consensus on symptomatic treatment,
- develop a foundation for longitudinal natural history studies, and
- illustrate, for the first time, that AP-4-HSP has features of both a neurodevelopmental and a neurodegenerative disease (Figure 3).
How Understanding AP-4-associated Protein Trafficking Provides Opportunities for Diagnosis and Treatment
With our clinical insights into AP-4-HSP, we turned to opportunities for diagnosis and treatment. Building on our collaborators’ groundbreaking work on AP-4 function, we discovered that AP-4’s role as an adaptor protein is to mediate the trafficking of transmembrane proteins from the trans-Golgi network to other compartments of the cell. AP-4 was found to be required for the correct trafficking of the autophagy-associated protein, ATG9A, and the endocannabinoid pathway enzyme, DAG lipase beta (DAGLB), among other less well characterized proteins associated with AP-4 vesicles.
In patient-derived fibroblasts and iPSC derived neurons, we found that ATG9A and DAGLB were mislocalized, leading to accumulation in the trans-Golgi network and depletion in other compartments of the cell, including axons. This strong cellular phenotype provided the basis for a diagnostic functional assay in human fibroblasts (Figure 4) and a starting point for the development of a phenotypic high-throughput screening platform. We have since used this assay to confirm diagnoses in cases with novel variants and atypical phenotypes. We have also completed several large small molecule and a first functional genomic screen that will provide key insights into the regulation of AP-4 mediated protein trafficking and therapeutic targets.
Moving Forward
Our work has grown beyond AP-4-HSP and we are working towards similar translational approaches to other rare genetic movement disorders. This includes gene discovery, the development of disease models in iPSCs and zebrafish, as well as high-throughput small molecule and functional genomics screens.
Future goals of our work include:
- To develop a broadly-accessible gene discovery platform to enable early and rapid genetic diagnoses in children with genetic movement disorders.
- To identify disease-associated mechanisms and develop next generation therapies.
- To build the infrastructure for first-in-human and later phase clinical trials in pediatric movement disorders.
We look forward to continuing our work with the IDDRC core facilities and to collaborating with other investigators in this center.
 Learn more about Dr. Ebrahimi-Fakhari
Learn more about Dr. Ebrahimi-Fakhari
We would like to thank the CureAP-4 foundation, created by the two families that originally met at BCH in supporting our research. We would also like to thank the NINDS, several other philanthropic organizations, as well as the IDDRC, the F.M. Kirby Neurology Center, and the Rosamund Stone Zander Translational Neuroscience Center, for allowing us to leverage the many resources available to us for moving our research forward.